


Quantum Mechanics and Molecular Dynamics Simulations
QM/MM simulation of polymer imprinting process
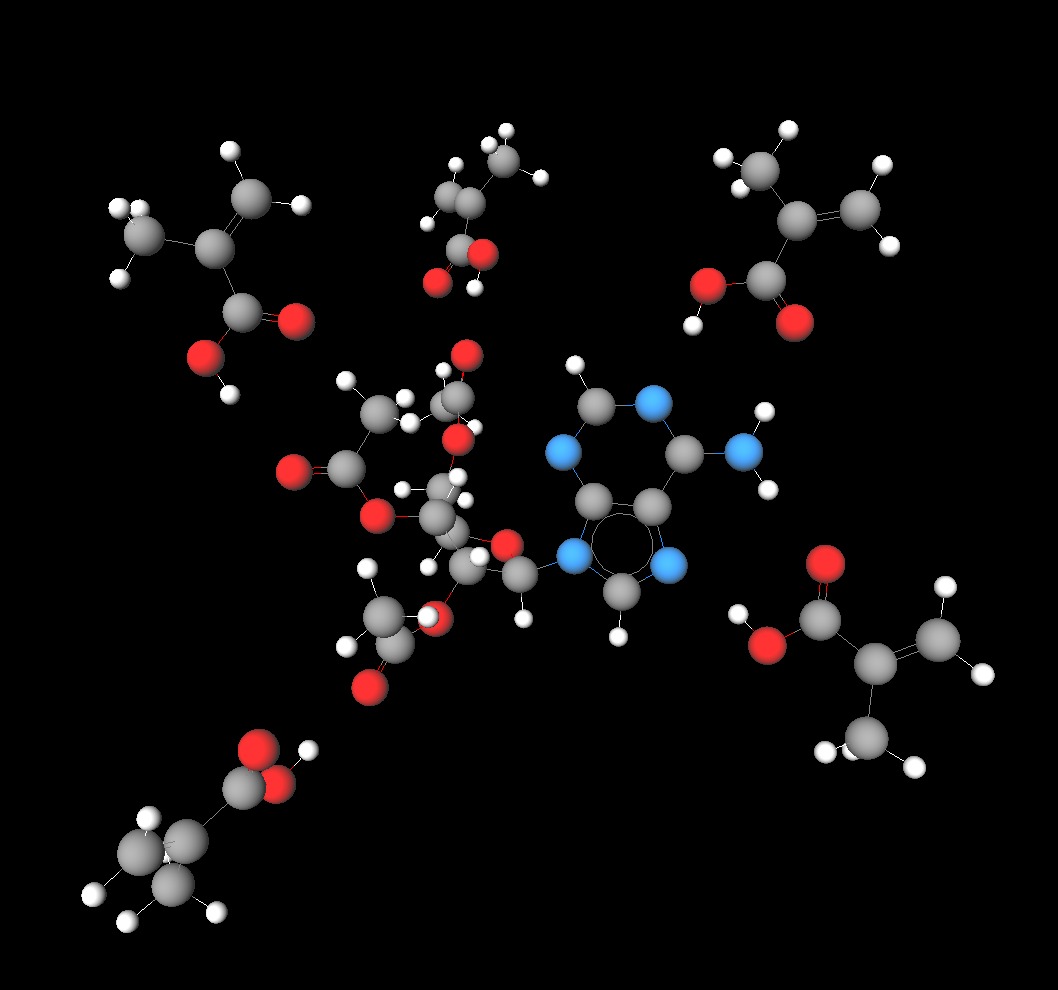
Tri-O-acetiladenosine molecule with five attached monomers of metacrylic acid.
Computer technologies and existing simulation methods make it possible to reduce the research costs for selecting imprinting parameters and estimate their optimal magnitudes in terms of such physicochemical criteria as template-monomer bond energy both in vacuum and solution, geometry and topology of the spatial structure of the pre-polymerization complex and the imprinted polymer, and their temperature dependencies.
The optimized geometrical structure and energy of the pre- polymerization complexes of tri-O-acetyladenosine (template) with three different monomers—methacrylic ac- id, 3-vinyl benzoic acid and acrylamide in vacuum were calculated using hybrid quantum mechanical/molecular me- chanical (QM/MM) approach. [Computer simulation based selection of optimal monomer for imprinting of tri-O-acetiladenosine in polymer matrix: vacuum calculations/Journal of Molecular Modelling, January 2013, Volume 19, Issue 1,pp. 359-369/
DOI 10.1007/s00894-012-1561-6/V.V. Barkaline,Y.Douhaya, A.Tsakalof]
Quantum mechanical study of the adsorption of gases on carbon nanotubes

Isosurfaces of electronic density corresponding to 0.01 a.u. from DFT calculation of hydrogen adsorption on carbon nanotube surface with cc-pvdz basis set (NW-Chem calculation and ECCE viewer).
Contact interaction of carbon nanotube with substrate

MD simulation of CNT contact interaction with smooth hydrogenized diamond surface
Molecular dynamic simulations of metals